
- Part Number:1910
- Part Number Title:Occupational Safety and Health Standards
- Subpart:1910 Subpart Z
- Subpart Title:Toxic and Hazardous Substances
- Standard Number:
- Title:Sampling and Analytical Methods for Ethylene Oxide (Non-Mandatory)
- GPO Source:
Appendix D to § 1910.1047 - Sampling and Analytical Methods for Ethylene Oxide (Non-Mandatory)
A number of methods are available for monitoring employee exposures to EtO. Most of these involve the use of charcoal tubes and sampling pumps, followed by analysis of the samples by gas chromatograph. The essential differences between the charcoal tube methods include, among others, the use of different desorbing solvents, the use of different lots of charcoal, and the use of different equipment for analysis of the samples.
Besides charcoal, methods using passive dosimeters, gas sampling bags, impingers, and detector tubes have been utilized for determination of EtO exposure. In addition, there are several commercially available portable gas analyzers and monitoring units.
This appendix contains details for the method which has been tested at the OSHA Analytical Laboratory in Salt Lake City. Inclusion of this method in the appendix does not mean that this method is the only one which will be satisfactory. Copies of descriptions of other methods available are available in the rulemaking record, and may be obtained from the OSHA Docket Office. These include the Union Carbide, Dow Chemical, 3M, and DuPont methods, as well as NIOSH Method S-286. These methods are briefly described at the end of this appendix.
Employers who note problems with sample breakthrough using the OSHA or other charcoal methods should try larger charcoal tubes. Tubes of larger capacity are available. In addition, lower flow rates and shorter sampling times should be beneficial in minimizing breakthrough problems. Whatever method the employer chooses, he must assure himself of the method's accuracy and precision under the unique conditions present in his workplace.
Ethylene Oxide
Method No.: 30.
Matrix: Air.
Target Concentration: 1.0 ppm (1.8 mg/m3).
Procedure: Samples are collected on two charcoal tubes in series and desorbed with 1% CS2 in benzene. The samples are derivatized with HBr and treated with sodium carbonate. Analysis is done by gas chromatography with an electron capture detector.
Recommended Air Volume and Sampling Rate: 1 liter and 0.05 Lpm.
Detection Limit of the Overall Procedure: 13.3 ppb (0.024 mg/m3) (Based on 1.0 liter air sample).
Reliable Quantitation Limit: 52.2 ppb (0.094 mg/m3) (Based on 1.0 liter air sample).
Standard Error of Estimate: 6.59% (See Backup Section 4.6).
Special Requirements: Samples must be analyzed within 15 days of sampling date.
Status of Method: The sampling and analytical method has been subjected to the established evaluation procedures of the Organic Method Evaluations Branch.
Date: August 1981.
Chemist: Wayne D. Potter.
Organic Solvents Branch, OSHA Analytical Laboratory, Salt Lake City, Utah
1. General Discussion.
1.1 Background.
1.1.1 History of Procedure.
Ethylene oxide samples analyzed at the OSHA Laboratory have normally been collected on activated charcoal and desorbed with carbon disulfide. The analysis is performed with a gas chromatograph equipped with a FID (Flame ionization detector) as described in NIOSH Method S286 (Ref. 5.1). This method is based on a PEL of 50 ppm and has a detection limit of about 1 ppm.
Recent studies have prompted the need for a method to analyze and detect ethylene oxide at very low concentrations.
Several attempts were made to form an ultraviolet (UV) sensitive derivative with ethylene oxide for analysis with HPLC. Among those tested that gave no detectable product were: p-anisidine, methylimidazole, aniline, and 2,3,6-trichlorobenzoic acid. Each was tested with catalysts such as triethylamine, aluminum chloride, methylene chloride and sulfuric acid but no detectable derivative was produced.
The next derivatization attempt was to react ethylene oxide with HBr to form 2-bromoethanol. This reaction was successful. An ECD (electron capture detector) gave a very good response for 2-bromoethanol due to the presence of bromine. The use of carbon disulfide as the desorbing solvent gave too large a response and masked the 2-bromoethanol. Several other solvents were tested for both their response on the ECD and their ability to desorb ethylene oxide from the charcoal. Among those tested were toluene, xylene, ethyl benzene, hexane, cyclohexane and benzene. Benzene was the only solvent tested that gave a suitable response on the ECD and a high desorption. It was found that the desorption efficiency was improved by using 1% CS2 with the benzene. The carbon disulfide did not significantly improve the recovery with the other solvents. SKC Lot 120 was used in all tests done with activated charcoal.
1.1.2 Physical Properties (Ref. 5.2-5.4).
Synonyms: Oxirane; dimethylene oxide, 1,2-epoxy-ethane; oxane; C2 H4 O; ETO;
Molecular Weight: 44.06
Boiling Point: 10.7 °C (51.3°)
Melting Point: −111 °C
Description: Colorless, flammable gas
Vapor Pressure: 1095 mm. at 20 °C
Odor: Ether-like odor
Lower Explosive Limits: 3.0% (by volume)
Flash Point (TOC): Below 0 °F
Molecular Structure: CH2 - CH2
1.2 Limit Defining Parameters.
1.2.1 Detection Limit of the Analytical Procedure.
The detection limit of the analytical procedure is 12.0 picograms of ethylene oxide per injection. This is the amount of analyte which will give a peak whose height is five times the height of the baseline noise. (See Backup Data Section 4.1).
1.2.2 Detection Limit of the Overall Procedure.
The detection limit of the overall procedure is 24.0 ng of ethylene oxide per sample.
This is the amount of analyte spiked on the sampling device which allows recovery of an amount of analyte equivalent to the detection limit of the analytical procedure. (See Backup Data Section 4.2).
1.2.3 Reliable Quantitation Limit.
The reliable quantitation limit is 94.0 nanograms of ethylene oxide per sample. This is the smallest amount of analyte which can be quantitated within the requirements of 75% recovery and 95% confidence limits. (See Backup Data Section 4.2).
It must be recognized that the reliable quantitation limit and detection limits reported in the method are based upon optimization of the instrument for the smallest possible amount of analyte. When the target concentration of an analyte is exceptionally higher than these limits, they may not be attainable at the routine operating parameters. In this case, the limits reported on analysis reports will be based on the operating parameters used during the analysis of the samples.
1.2.4 Sensitivity.
The sensitivity of the analytical procedure over a concentration range representing 0.5 to 2 times the target concentration based on the recommended air volume is 34105 area units per µg/mL. The sensitivity is determined by the slope of the calibration curve (See Backup Data Section 4.3).
The sensitivity will vary somewhat with the particular instrument used in the analysis.
1.2.5 Recovery.
The recovery of analyte from the collection medium must be 75% or greater. The average recovery from spiked samples over the range of 0.5 to 2 times the target concentration is 88.0% (See Backup Section 4.4). At lower concentrations the recovery appears to be non-linear.
1.2.6 Precision (Analytical Method Only).
The pooled coefficient of variation obtained from replicate determination of analytical standards at 0.5X, 1X and 2X the target concentration is 0.036 (See Backup Data Section 4.5).
1.2.7 Precision (Overall Procedure).
The overall procedure must provide results at the target concentration that are 25% of better at the 95% confidence level. The precision at the 95% confidence level for the 15 day storage test is plus or minus 12.9% (See Backup Data Section 4.6).
This includes an additional plus or minus 5% for sampling error.
1.3 Advantages.
1.3.1 The sampling procedure is convenient.
1.3.2 The analytical procedure is very sensitive and reproducible.
1.3.3 Reanalysis of samples is possible.
1.3.4 Samples are stable for at least 15 days at room temperature.
1.3.5 Interferences are reduced by the longer GC retention time of the new derivative.
1.4 Disadvantages.
1.4.1 Two tubes in series must be used because of possible breakthrough and migration.
1.4.2 The precision of the sampling rate may be limited by the reproducibility of the pressure drop across the tubes. The pumps are usually calibrated for one tube only.
1.4.3 The use of benzene as the desorption solvent increases the hazards of analysis because of the potential carcinogenic effects of benzene.
1.4.4 After repeated injections there can be a buildup of residue formed on the electron capture detector which decreases sensitivity.
1.4.5 Recovery from the charcoal tubes appears to be nonlinear at low concentrations.
2. Sampling Procedure.
2.1 Apparatus.
2.1.1 A calibrated personal sampling pump whose flow can be determined within plus or minus 5% of the recommended flow.
2.1.2 SKC Lot 120 Charcoal tubes: glass tube with both ends flame sealed, 7 cm long with a 6 mm O.D. and a 4-mm I.D., containing 2 sections of coconut shell charcoal separated by a 2-mm portion of urethane foam. The adsorbing section contains 100 mg of charcoal, the backup section 50 mg. A 3-mm portion of urethane foam is placed between the outlet end of the tube and the backup section. A plug of silylated glass wool is placed in front of the adsorbing section.
2.2 Reagents.
2.2.1 None required.
2.3 Sampling Technique.
2.3.1 Immediately before sampling, break the ends of the charcoal tubes. All tubes must be from the same lot.
2.3.2 Connect two tubes in series to the sampling pump with a short section of flexible tubing. A minimum amount of tubing is used to connect the two sampling tubes together. The tube closer to the pump is used as a backup. This tube should be identified as the backup tube.
2.3.3 The tubes should be placed in a vertical position during sampling to minimize channeling.
2.3.4 Air being sampled should not pass through any hose or tubing before entering the charcoal tubes.
2.3.5 Seal the charcoal tubes with plastic caps immediately after sampling. Also, seal each sample with OSHA seals lengthwise.
2.3.6 With each batch of samples, submit at least one blank tube from the same lot used for samples. This tube should be subjected to exactly the same handling as the samples (break, seal, transport) except that no air is drawn through it.
2.3.7 Transport the samples (and corresponding paperwork) to the lab for analysis.
2.3.8 If bulk samples are submitted for analysis, they shoud be transported in glass containers with Teflon-lined caps. These samples must be mailed separately from the container used for the charcoal tubes.
2.4 Breakthrough.
2.4.1 The breakthrough (5% breakthrough) volume for a 3.0 mg/m ethylene oxide sample stream at approximately 85% relative humidity, 22 °C and 633 mm is 2.6 liters sampled at 0.05 liters per minute. This is equivalent to 7.8 µg of ethylene oxide. Upon saturation of the tube it appeared that the water may be displacing ethylene oxide during sampling.
2.5 Desorption Efficiency.
2.5.1 The desorption efficiency, from liquid injection onto charcoal tubes, averaged 88.0% from 0.5 to 2.0 × the target concentration for a 1.0 liter air sample. At lower ranges it appears that the desorption efficiency is non-linear (See Backup Data Section 4.2).
2.5.2 The desorption efficiency may vary from one laboratory to another and also from one lot of charcoal to another. Thus, it is necessary to determine the desorption efficiency for a particular lot of charcoal.
2.6 Recommended Air Volume and Sampling Rate.
2.6.1 The recommended air volume is 1.0 liter.
2.6.2 The recommended maximum sampling rate is 0.05 Lpm.
2.7 Interferences.
2.7.1 Ethylene glycol and Freon 12 at target concentration levels did not interfere with the collection of ethylene oxide.
2.7.2 Suspected interferences should be listed on the sample data sheets.
2.7.3 The relative humidity may affect the sampling procedure.
2.8 Safety Precautions.
2.8.1 Attach the sampling equipment to the employee so that it does not interfere with work performance.
2.8.2 Wear safety glasses when breaking the ends of the sampling tubes.
2.8.3 If possible, place the sampling tubes in a holder so the sharp end is not exposed while sampling.
3. Analytical Method.
3.1 Apparatus.
3.1.1 Gas chromatograph equipped with a linearized electron capture detector.
3.1.2 GC column capable of separating the derivative of ethylene oxide (2-bromoethanol) from any interferences and the 1% CS2 in benzene solvent. The column used for validation studies was: 10 ft × ⅛ inch stainless steel 20% SP-2100, .1% Carbowax 1500 on 100/120 Supelcoport.
3.1.3 An electronic integrator or some other suitable method of measuring peak areas.
3.1.4 Two milliliter vials with Teflon-lined caps.
3.1.5 Gas tight syringe - 500 µL or other convenient sizes for preparing standards.
3.1.6 Microliter syringes - 10 µL or other convenient sizes for diluting standards and 1 µL for sample injections.
3.1.7 Pipets for dispensing the 1% CS2 in benzene solvent. The Glenco 1 mL dispenser is adequate and convenient.
3.1.8 Volumetric flasks - 5 mL and other convenient sizes for preparing standards.
3.1.9 Disposable Pasteur pipets.
3.2 Reagents.
3.2.1 Benzene, reagent grade.
3.2.2 Carbon Disulfide, reagent grade.
3.2.3 Ethylene oxide, 99.7% pure.
3.2.4 Hydrobromic Acid, 48% reagent grade.
3.2.5 Sodium Carbonate, anhydrous, reagent grade.
3.2.6 Desorbing reagent, 99% Benzene/1% CS2.
3.3 Sample Preparation.
3.3.1 The front and back sections of each sample are transferred to separate 2-mL vials.
3.3.2 Each sample is desorbed with 1.0 mL of desorbing reagent.
3.3.3 The vials are sealed immediately and allowed to desorb for one hour with occasional shaking.
3.3.4 Desorbing reagent is drawn off the charcoal with a disposable pipet and put into clean 2-mL vials.
3.3.5 One drop of HBr is added to each vial. Vials are resealed and HBr is mixed well with the desorbing reagent.
3.3.6 About 0.15 gram of sodium carbonate is carefully added to each vial. Vials are again resealed and mixed well.
3.4 Standard Preparation.
3.4.1 Standards are prepared by injecting the pure ethylene oxide gas into the desorbing reagent.
3.4.2 A range of standards are prepared to make a calibration curve. A concentration of 1.0 µL of ethylene oxide gas per 1 mL desorbing reagent is equivalent to 1.0 ppm air concentration (all gas volumes at 25 °C and 760 mm) for the recommended 1 liter air sample. This amount is uncorrected for desorption efficiency (See Backup Data Section 4.2. for desorption efficiency corrections).
3.4.3 One drop of HBr per mL of standard is added and mixed well.
3.4.4 About 0.15 grams of sodium carbonate is carefully added for each drop of HBr (A small reaction will occur).
3.5 Analysis.
3.5.1 GC Conditions.
Nitrogen flow rate - 10mL/min.
Injector Temperature - 250 °C
Detector Temperature - 300 °C
Column Temperature - 100 °C
Injection size - 0.8 µL
Elution time - 3.9 minutes
3.5.2 Peak areas are measured by an integrator or other suitable means.
3.5.3 The integrator results are in area units and a calibration curve is set up with concentration vs. area units.
3.6 Interferences.
3.6.1 Any compound having the same retention time of 2-bromoethanol is a potential interference. Possible interferences should be listed on the sample data sheets.
3.6.2 GC parameters may be changed to circumvent interferences.
3.6.3 There are usually trace contaminants in benzene. These contaminants, however, posed no problem of interference.
3.6.4 Retention time data on a single column is not considered proof of chemical identity. Samples over the 1.0 ppm target level should be confirmed by GC/Mass Spec or other suitable means.
3.7 Calculations
3.7.1 The concentration in µg/mL for a sample is determined by comparing the area of a particular sample to the calibration curve, which has been prepared from analytical standards.
3.7.2 The amount of analyte in each sample is corrected for desorption efficiency by use of a desorption curve.
3.7.3 Analytical results (A) from the two tubes that compose a particular air sample are added together.
3.7.4 The concentration for a sample is calculated by the following equation:
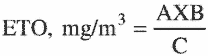
where:
A = µg/mL
B = desorption volume in milliliters
C = air volume in liters.
3.7.5 To convert mg/m3 to parts per million (ppm) the following relationship is used:
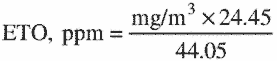
where:
mg/m3 = results from 3.7.4
24.45 = molar volume at 25 °C and 760mm Hg
44.05 = molecular weight of ETO.
3.8 Safety Precautions
3.8.1 Ethylene oxide and benzene are potential carcinogens and care must be exercised when working with these compounds.
3.8.2 All work done with the solvents (preparation of standards, desorption of samples, etc.) should be done in a hood.
3.8.3 Avoid any skin contact with all of the solvents.
3.8.4 Wear safety glasses at all times.
3.8.5 Avoid skin contact with HBr because it is highly toxic and a strong irritant to eyes and skin.
4. Backup Data.
4.1 Detection Limit Data.
The detection limit was determined by injecting 0.8 µL of a 0.015 µg/mL standard of ethylene oxide into 1% CS2 in benzene. The detection limit of the analytical procedure is taken to be 1.20 × 10−5 µg per injection. This is equivalent to 8.3 ppb (0.015 mg/m3) for the recommended air volume.
4.2 Desorption Efficiency.
Ethylene oxide was spiked onto charcoal tubes and the following recovery data was obtained.
| Amount spiked (µg) | Amount recovered (µg) | Percent recovery |
|---|---|---|
| 4.5 | 4.32 | 96.0 |
| 3.0 | 2.61 | 87.0 |
| 2.25 | 2.025 | 90.0 |
| 1.5 | 1.365 | 91.0 |
| 1.5 | 1.38 | 92.0 |
| .75 | .6525 | 87.0 |
| .375 | .315 | 84.0 |
| .375 | .312 | 83.2 |
| .1875 | .151 | 80.5 |
| .094 | .070 | 74.5 |
At lower amounts the recovery appears to be non-linear.
4.3 Sensitivity Data.
The following data was used to determine the calibration curve.
| Injection | 0.5 × .75 µg/mL | 1 × 1.5 µg/mL | 2 × 3.0 µg/mL |
|---|---|---|---|
| 1 | 30904 | 59567 | 111778 |
| 2 | 30987 | 62914 | 106016 |
| 3 | 32555 | 58578 | 106122 |
| 4 | 32242 | 57173 | 109716 |
| X | 31672 | 59558 | 108408 |
Slope = 34.105.
4.4 Recovery.
The recovery was determined by spiking ethylene oxide onto lot 120 charcoal tubes and desorbing with 1% CS2 in Benzene. Recoveries were done at 0.5, 1.0, and 2.0× the target concentration (1 ppm) for the recommended air volume.
| Sample | 0.5x | 1.0x | 2.0x |
|---|---|---|---|
| 1 | 88.7 | 95.0 | 91.7 |
| 2 | 83.8 | 95.0 | 87.3 |
| 3 | 84.2 | 91.0 | 86.0 |
| 4 | 88.0 | 91.0 | 83.0 |
| 5 | 88.0 | 86.0 | 85.0 |
| X | 86.5 | 90.5 | 87.0 |
Weighted Average = 88.2.
4.5 Precision of the Analytical Procedure.
The following data was used to determine the precision of the analytical method:
| Concentration | 0.5 × .75 µg/mL | 1 × 1.5 µg/mL | 2 × 3.0 µg/mL |
|---|---|---|---|
| Injection | .7421 | 1.4899 | 3.1184 |
| .7441 | 1.5826 | 3.0447 | |
| .7831 | 1.4628 | 2.9149 | |
| .7753 | 1.4244 | 2.9185 | |
| Average | .7612 | 1.4899 | 2.9991 |
| Standard Deviation | .0211 | .0674 | .0998 |
| CV | .0277 | .0452 | .0333 |

CV + 0.036
4.6 Storage Data.
Samples were generated at 1.5 mg/m3 ethylene oxide at 85% relative humidity, 22 °C and 633 mm. All samples were taken for 20 minutes at 0.05 Lpm. Six samples were analyzed as soon as possible and fifteen samples were stored at refrigerated temperature (5 °C) and fifteen samples were stored at ambient temperature (23 °C). These stored samples were analyzed over a period of nineteen days.
| Day analyzed | Refrigerated | Ambient |
|---|---|---|
| 1 | 87.0 | 87.0 |
| 1 | 93.0 | 93.0 |
| 1 | 94.0 | 94.0 |
| 1 | 92.0 | 92.0 |
| 4 | 92.0 | 91.0 |
| 4 | 93.0 | 88.0 |
| 4 | 91.0 | 89.0 |
| 6 | 92.0 | |
| 6 | 92.0 | |
| 8 | 92.0 | |
| 8 | 86.0 | |
| 10 | 91.7 | |
| 10 | 95.5 | |
| 10 | 95.7 | |
| 11 | 90.0 | |
| 11 | 82.0 | |
| 13 | 78.0 | |
| 13 | 81.4 | |
| 13 | 82.4 | |
| 14 | 78.5 | |
| 14 | 72.1 | |
| 18 | 66.0 | |
| 18 | 68.0 | |
| 19 | 64.0 | |
| 19 | 77.0 |
4.7 Breakthrough Data.
Breakthrough studies were done at 2 ppm (3.6 mg/m3) at approximately 85% relative humidity at 22 °C (ambient temperature). Two charcoal tubes were used in series. The backup tube was changed every 10 minutes and analyzed for breakthrough. The flow rate was 0.050 Lpm.
| Tube No. | Time (minutes) | Percent breakthrough |
|---|---|---|
| 1 | 10 | (1) |
| 2 | 20 | (1) |
| 3 | 30 | (1) |
| 4 | 40 | 1.23 |
| 5 | 50 | 3.46 |
| 6 | 60 | 18.71 |
| 7 | 70 | 39.2 |
| 8 | 80 | 53.3 |
| 9 | 90 | 72.0 |
| 10 | 100 | 96.0 |
| 11 | 110 | 113.0 |
| 12 | 120 | 133.9 |
1 None.
The 5% breakthrough volume was reached when 2.6 liters of test atmosphere were drawn through the charcoal tubes.
5. References.
5.1 “NIOSH Manual of Analytical Methods,” 2nd ed. NIOSH: Cincinnati, 1977; Method S286.
5.2 “IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man,” International Agency for Research on Cancer: Lyon, 1976; Vol. II, p. 157.
5.3 Sax., N.I. “Dangerous Properties of Industrial Materials,” 4th ed.; Van Nostrand Reinhold Company. New York, 1975; p. 741.
5.4 “The Condensed Chemical Dictionary”, 9th ed.; Hawley, G.G., ed.; Van Nostrand Reinhold Company, New York, 1977; p. 361.
Summary of Other Sampling Procedures
OSHA believes that served other types of monitoring equipment and techniques exist for monitoring time-weighted averages. Considerable research and method development is currently being performed, which will lead to improvements and a wider variety of monitoring techniques. A combination of monitoring procedures can be used. There probably is no one best method for monitoring personal exposure to ethylene oxide in all cases. There are advantages, disadvantages, and limitations to each method. The method of choice will depend on the need and requirements. Some commonly used methods include the use of charcoal tubes, passive dosimeters, Tedler gas sampling bags, detector tubes, photoionization detection units, infrared detection units and gas chromatographs. A number of these methods are described below.
A. Charcoal Tube Sampling Procedures
Qazi-Ketcham method (Ex. 11-133) - This method consists of collecting EtO on Columbia JXC activated carbon, desorbing the EtO with carbon disulfide and analyzing by gas chromatography with flame ionization detection. Union Carbide has recently updated and revalidated this monitoring procedures. This method is capable of determining both eight-hour time-weighted average exposures and short-term exposures. The method was validated to 0.5 ppm. Like other charcoal collecting procedures, the method requires considerable analytical expertise.
ASTM-proposed method - The Ethylene Oxide Industry Council (EOIC) has contracted with Clayton Environmental Consultants, Inc. to conduct a collaborative study for the proposed method. The ASTM-Proposed method is similar to the method published by Qazi and Ketcham is the November 1977 American Industrial Hygiene Association Journal, and to the method of Pilney and Coyne, presented at the 1979 American Industrial Hygiene Conference. After the air to be sampled is drawn through an activated charcoal tube, the ethylene oxide is desorbed from the tube using carbon disulfide and is quantitated by gas chromatography utilizing a flame ionization detector. The ASTM-proposed method specifies a large two-section charcoal tube, shipment in dry ice, storage at less than −5 °C, and analysis within three weeks to prevent migration and sample loss. Two types of charcoal tubes are being tested - Pittsburgh Coconut-Based (PCB) and Columbia JXC charcoal. This collaborative study will give an indication of the inter- and intralaboratory precision and accuracy of the ASTM-proposed method. Several laboratories have considerable expertise using the Qazi-Ketcham and Dow methods.
B. Passive Monitors - Ethylene oxide diffuses into the monitor and is collected in the sampling media. The DuPont Pro-Tek badge collects EtO in an absorbing solution, which is analyzed colorimetrically to determine the amount of EtO present. The 3M 350 badge collects the EtO on chemically treated charcoal. Other passive monitors are currently being developed and tested. Both 3M and DuPont have submitted data indicating their dosimeters meet the precision and accuracy requirements of the proposed ethylene oxide standard. Both presented laboratory validation data to 0.2 ppm (Exs. 11-65, 4-20, 108, 109, 130).
C. Tedlar Gas Sampling Bags-Samples are collected by drawing a known volume of air into a Tedlar gas sampling bag. The ethylene oxide concentration is often determined on-site using a portable gas chromatograph or portable infrared spectometer.
D. Detector tubes - A known volume of air is drawn through a detector tube using a small hand pump. The concentration of EtO is related to the length of stain developed in the tube. Detector tubes are economical, easy to use, and give an immediate readout. Unfortunately, partly because they are nonspecific, their accuracy is often questionable. Since the sample is taken over a short period of time, they may be useful for determining the source of leaks.
E. Direct Reading Instruments - There are numerous types of direct reading instruments, each having its own strengths and weaknesses (Exs. 135B, 135C, 107, 11-78, 11-153). Many are relatively new, offering greater sensitivity and specificity. Popular ethylene oxide direct reading instruments include infrared detection units, photoionization detection units, and gas chromatographs.
Portable infrared analyzers provide an immediate, continuous indication of a concentration value; making them particularly useful for locating high concentration pockets, in leak detection and in ambient air monitoring. In infrared detection units, the amount of infrared light absorbed by the gas being analyzed at selected infrared wavelengths is related to the concentration of a particular component. Various models have either fixed or variable infrared filters, differing cell pathlengths, and microcomputer controls for greater sensitivity, automation, and interference elimination.
A fairly recent detection system is photoionization detection. The molecules are ionized by high energy ultraviolet light. The resulting current is measured. Since different substances have different ionization potentials, other organic compounds may be ionized. The lower the lamp energy, the better the selectivity. As a continuous monitor, photoionization detection can be useful for locating high concentration pockets, in leak detection, and continuous ambient air monitoring. Both portable and stationary gas chromatographs are available with various types of detectors, including photoionization detectors. A gas chromatograph with a photoionization detector retains the photionization sensitivity, but minimizes or eliminates interferences. For several GC/PID units, the sensitivity is in the 0.1-0.2 ppm EtO range. The GC/PID with microprocessors can sample up to 20 sample points sequentially, calculate and record data, and activate alarms or ventilation systems. Many are quite flexible and can be configured to meet the specific analysis needs for the workplace.
DuPont presented their laboratory validation data of the accuracy of the Qazi-Ketcham charcoal tube, the PCB charcoal tube, Miran 103 IR analyzer, 3M #3550 monitor and the Du Pont C-70 badge. Quoting Elbert V. Kring:
We also believe that OSHA's proposed accuracy in this standard is appropriate. At plus or minus 25 percent at one part per million, and plus or minus 35 percent below that. And, our data indicates there's only one monitoring method, right now, that we've tested thoroughly, that meets that accuracy requirements. That is the Du Pont Pro-Tek badge* * *. We also believe that this kind of data should be confirmed by another independent laboratory, using the same type dynamic chamber testing (Tr. 1470)
Additional data by an independent laboratory following their exact protocol was not submitted. However, information was submitted on comparisons and precision and accuracy of those monitoring procedures which indicate far better precision and accuracy of those monitoring procedures than that obtained by Du Pont (Ex. 4-20, 130, 11-68, 11-133, 130, 135A).
The accuracy of any method depends to a large degree upon the skills and experience of those who not only collect the samples but also those who analyze the samples. Even for methods that are collaboratively tested, some laboratories are closer to the true values than others. Some laboratories may meet the precision and accuracy requirements of the method; others may consistently far exceed them for the same method.
[49 FR 25796, June 22, 1984, as amended at 50 FR 9801, Mar. 12, 1985; 50 FR 41494, Oct. 11, 1985; 51 FR 25053, July 10, 1986; 53 FR 11436, 11437, Apr. 6, 1988; 53 FR 27960, July 26, 1988; 54 FR 24334, June 7, 1989; 61 FR 5507, Feb. 13, 1996]